Esfandiarpour, A, and Nasrabadi, M.N. “Vacancy Formation Energy in CuNiCo Equimolar Alloy and CuNiCoFe High Entropy Alloy: Ab Initio Based Study.” Calphad, vol. 66, 2019, p. 101634.
Kivy, Mohsen & Hong, Yu & Asle Zaeem, Mohsen. (2019). A Review of Multi-Scale Computational Modeling Tools for Predicting Structures and Properties of Multi-Principal Element Alloys. Metals. 9. 254. 10.3390/met9020254.
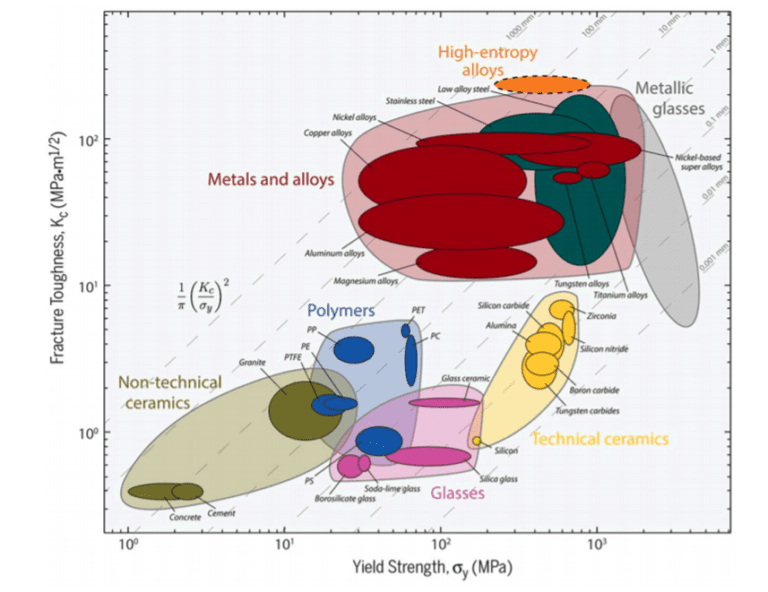
Miracle, D.B, and Senkov, O.N. “A Critical Review of High Entropy Alloys and Related Concepts.” Acta 2019, nanohub.org/resources/sqsatat/aboutMaterialia, vol. 122, 2017, pp. 448–511.
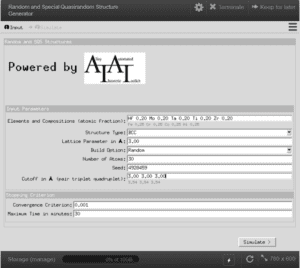
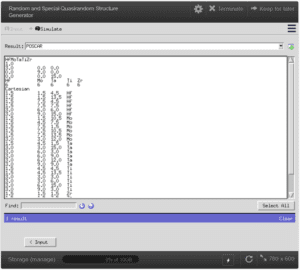
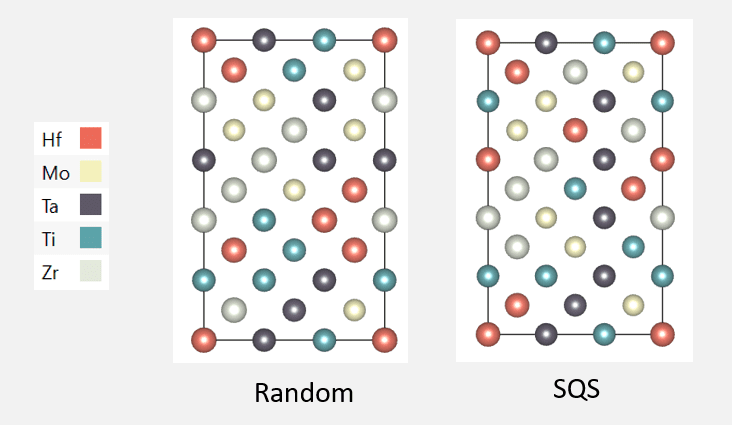
Tripathi, Shivam, et al. “Random and Special Quasirandom Structure Generator.” NanoHUB.org, NCN, 30 Sept. 2019.


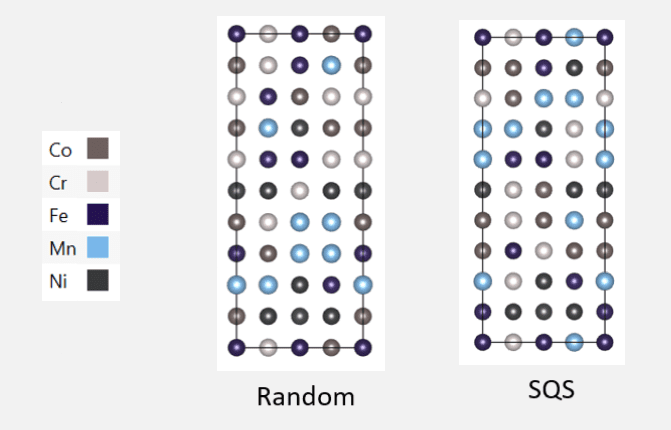
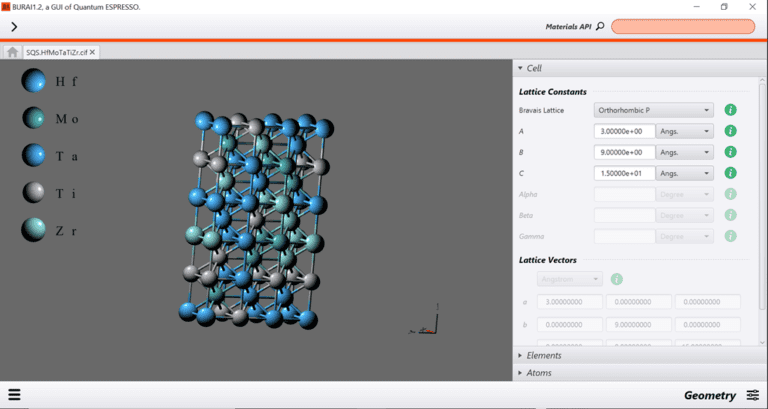
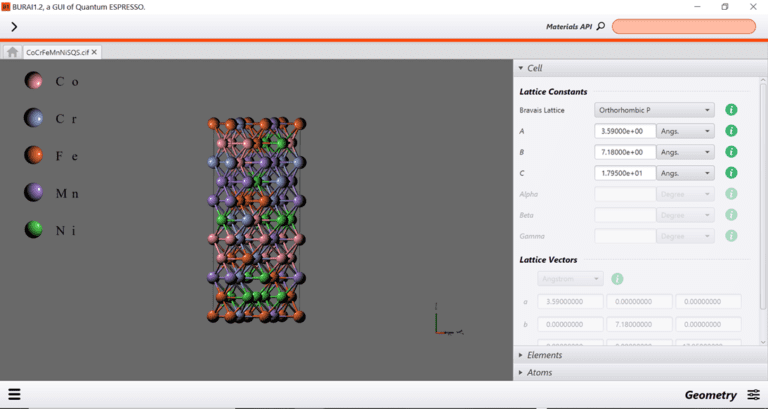
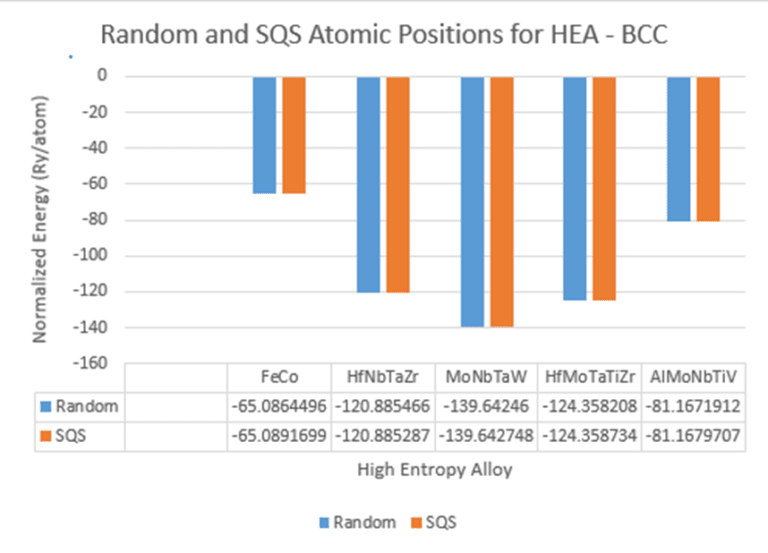
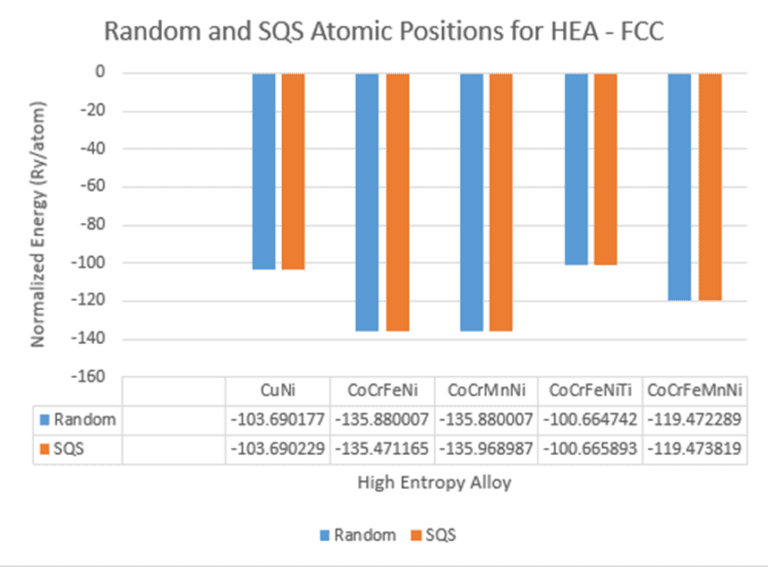
{kind=link}
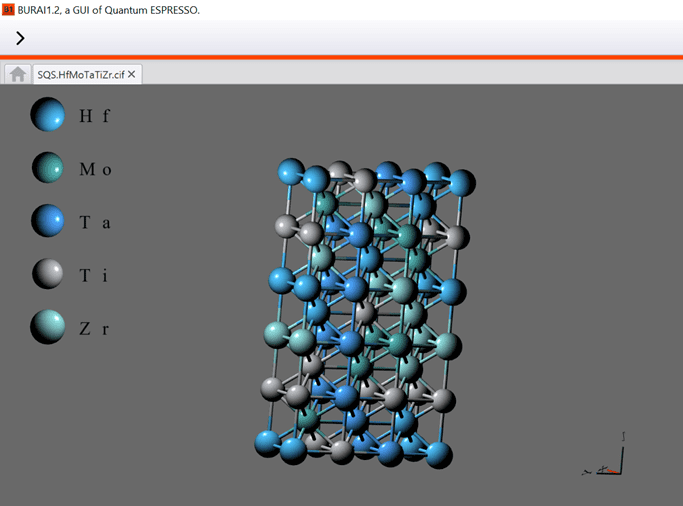
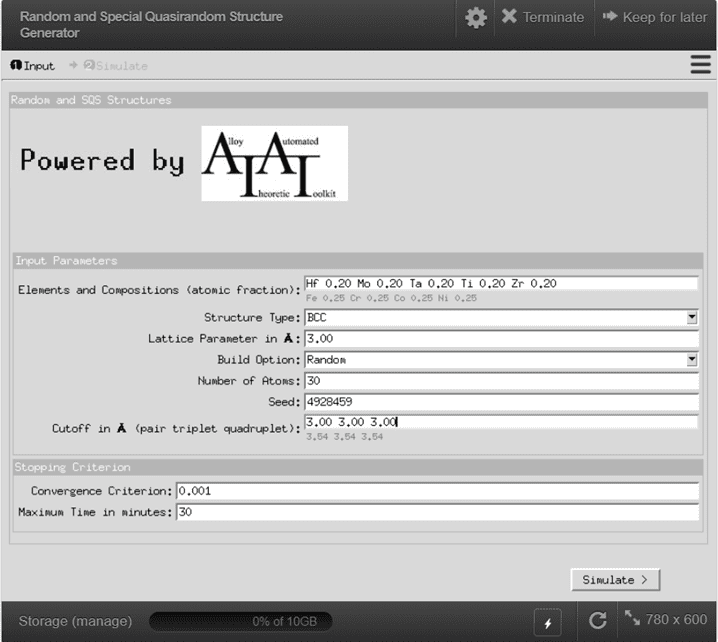
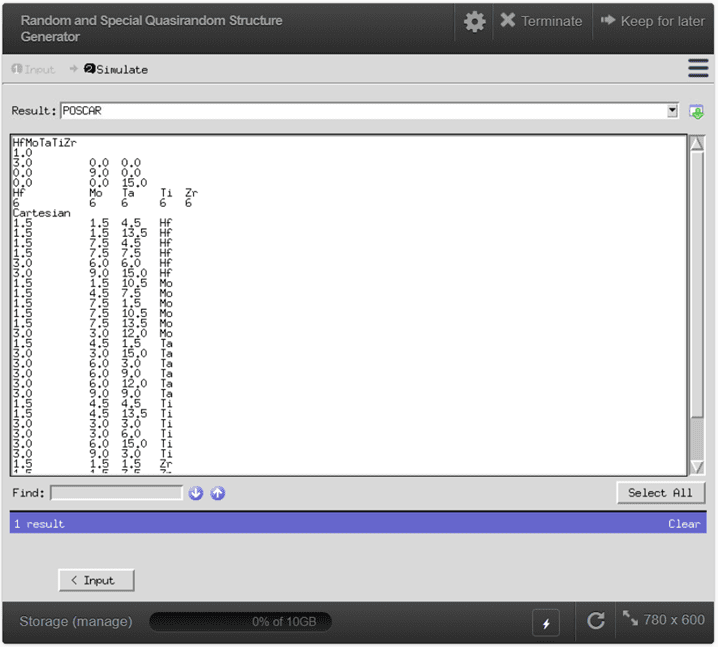{kind=link}
{kind=link}
{kind=link}